|
Optic
nerve gliomas are rare, comprising 1% of all intracranial tumors; but
4-6% of all brain tumors in children. Eighty-five percent occur in
children under 15 years of age and the female/male ratio is 3:2.
Pathology:
About
10 per cent of optic pathway tumors are located within an optic nerve.
One third of the tumors involve both optic nerve and chiasm, a further
third involve predominantly the chiasm itself, and one fourth are
predominantly in the hypothalamus.5 5% are multicentric.
Although
it has previously been suggested that these lesions are hamartomas, most
authorities now accept that they are truly neoplastic. These tumors are
usually slow-growing low grade (pilocytic) gliomas. Occasionally
malignant ones also found at this location usually in adults. Rarely, a
malignant optic glioma can occur in a child. In childhood, the optic
pathway tumors are low-grade astrocytomas and rarely gangliogliomas.
However, optic pathway gliomas can occur in adults, and in that situation
the tumors may have the characteristics of a highly malignant tumor that
causes early loss of vision and inevitably leads to the patient's death.
Macroscopically,
these tumors may be solid, gelatinous or cystic. Although having certain
gross similarities with oligodendrocytes, closer microscopic,
ultrastructural and immunostaining techniques have confirmed their low
grade astrocytic (pilocytic) nature with the presence of numerous
Rosenthal’s fibres. The tumor may start in the anterior end of the optic
nerve and proceed backwards intracranially or may arise originally from
the optic nerve-chiasma junction. Occasionally, a glioma from the optic
tract or the anterior third ventricle region may involve the chiasma and
the optic nerve secondarily. About 40 per cent of optic pathway
astrocytomas are fibrillary and 60 per cent are pilocytic. Hypothalamic
tumors which have invaded the optic chiasm behave differently, showing
evidence of local invasion and histologically are not pilocytic in nature
but are similar to other cerebral hemisphere gliomas.
About
one third of the patients with optic pathway hypothalamic gliomas will
have neurofibromatosis type1. Patients with neurofibromatosis have
histologically similar tumors but the tumors are more likely to be
associated with extensive arachnoidal hyperplasia and florid local
gliomatous infiltration of the leptomeninges. Indeed, in NF-1 patients
optic nerve lesions may be due to diffuse hyperplasitc gliosis rather
than actual neoplasia. Multicentric tumors involving both optic nerves
are also usually associated with NF-1.
Clinical
features:
The
evolution of symptoms is slow and insidious and depends on the site of
origin. In general, optic nerve tumors present at a slightly later age (6
years) than the hypothalamic/chiasmatic tumors (2 to 4 years).
Tumors
restricted to optic nerve produce failing vision in one eye. It is often
not detected by children for a long time. Papilledema (probably due to
venous obstruction), being more common than optic atrophy is the cause
for visual failure. Proptosis is delayed. Optic atrophy in a child with
no retinal or macular degeneration points to an optic nerve glioma,
especially in neurofibromatosis. 20-30% of the patients have café-au-lait
spots. Irregular visual field defect is diagnostic. Initially there is
central or paracentral scotoma.
Tumors
that involve the chiasma produce bilateral irregular visual field defect.
The loss of visual acuity is secondary to optic atrophy.
Unfortunately
posteriorly located tumors are the most common form of optic nerve
gliomas (60%) and it is often difficult to determine the actual origin of
the tumor as they are frequently large and involve both the chiasm and
the hypothalamus. Patients may present with hydrocephalus (due to obstruction
of the foramen of Monro), visual problems (acuity and field defects),
pituitary dysfunction or hypothalamic dysfunction. The latter,
classically leading to the diencephalic syndrome (emaciation, pallor, and
hyperactivity), is seen in up to 20% of patients under 3 years of age.
When the hypothalamus is involved, particularly in infancy, the child may
present with a diencephalic syndrome.3 Other symptoms of
hypothalamic involvement include diabetes insipidus, anorexia, obesity
and precocious puberty.
Differential
diagnosis:
Optic
nerve glioma
Hemangioma, Lymphoma, Rhabdomyosarcoma, Metastases (Neuroblastoma,
Leukamia, Ewing’s sarcoma), Fibrous dysplasia, Paranasal mucocoele,
Meningioma, Neurofibromatosis
(Orbital neurofibroma, Congenital defect in sphenoid bone)
Optic
nerve and chiasm glioma
Germinoma, Sarcoidosis
Optic
chiasm glioma extending into the hypothalamus
Pituitary adenoma, Craniopharyngioma, Malignant
astrocytoma, Epidermoid
and dermoids, Chordoma, Colloid cyst, Fibrous dyplasia, Sarcoidosis,
Histiocytosis X
Tuberculous granuloma, Hemangloendothelioma.
|
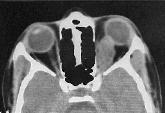
Investigations:
Classical
skull X-ray changes (widening of the optic foramen, shaped sella) have
been superseded by CT and MRI. Bone window setting on CT often reveals
widening of the optic canal. The optic nerve tumor is usually lobulated
but may be smooth and fusiform. It almost always enhances after
contrast. Intracranial tumors appear as low-density lobulated masses on
CT and show inhomogeneous contract enhancement.
|
|
|
|
|
Optic nerve glioma-CT
|
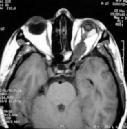
Optic nerve glioma-MRI
|
|
MRI
is far more sensitive for displaying chiasmatic/hypothalamic tumors than
CT. These tumors are usually hypointense on T1-weighted images and
hyperintense on T2 and almost always, enhance with gadolinium. On T2,
high intensity signal may be seen extending to the lateral geniculate
bodies.
When
the tumor involves the chiasm and the hypothalamus, it can reach an
enormous size, particularly in infancy. In such situations there is often
extensive distention of the basal cisterns, which can rupture, giving
rise to large subdural hygromas that can envelop both cerebral
hemispheres.
Visual
evoked responses may be of assistance in monitoring visual function; but
it is of limited value.
Management:
They
are usually slow growing and very occasionally may become malignant and
grow rapidly. Ten year survival rates also depend on site and range from 76%
to 95% for posteriorly and anteriorly situated pilocytic tumors
respectively. The treatment remains controversial and is dependent upon
the site of the tumor especially when the optic chiasm and/or the
hypothalamus is involved, and ranges from observation alone to operative
intervention with or without adjuvant therapy. Some physicians advocate
resection without any further therapy, others recommend radiation therapy
or chemotherapy, and still others believe that no therapy is required.
The
natural history of this tumor is variable and spontaneous regression can
occur. This should be kept in mind in deciding on treatment. Patients
with an optic glioma and neurofibromatosis type 1 have a better prognosis
than those who do not have neurofibromatosis.
Orbital
and Intracranial optic nerve tumors: Not infrequently, asymptomatic optic
nerve tumors are detected whilst imaging patients with neurofibromatosis.
In those patients with reasonable and/or static visual acuity,
surveillance with regular ophthalmological assessment and imaging may be
appropriate.
The
aim of surgical treatment is to remove the tumor before there has been
spread to the optic chiasm or contralateral optic nerve and to deal with
the associated proptosis. Although in patients with orbital optic nerve
tumors this may be achieved with relatively little morbidity via an
extracranial resection (transorbital approach), the risk of leaving tumor
in the optic nerve stump has resulted in most surgeons treating these
lesions in the same fashion as those that involve the intracranial optic
nerve – with excision of the optic nerve just distal to the optic chiasm
to avoid sectioning through the looping fibers of Willbrand.
Patients with an optic glioma and neurofibromatosis type 1 have a better
prognosis than those who do not have neurofibromatosis. An orbital glioma
can be removed in two pieces. The intracranial optic nerve is removed
from in front of the chiasm to the optic canal, and the orbital glioma is
removed from the globe to the optic canal. The residual intracanalicular
tumor is then coagulated. By doing this there is no need to section the
annulus or to divide the origin of the levator and possibly damage the
trochlear nerve.
Optic
nerve and chiasmatic tumors: The number of patients reported in the
literature is small; it would appear that long-term survival should be
expected in this group. Surgery is reserved for diagnostic purposes or to
excise an exophytic component: adjunctive therapy is reserved for tumor
progression.
Chiasmatic/hypothalamic
tumors: These tumors show markedly varying capacity for progression with
some remaining indolent while others rapidly increase in size. Operative
debulking of the tumor mass with or without adjuvant therapy is utilized
in cases of tumor progression. Although long-term survival has been
reported with these tumors, this is usually accompanied by significant
morbidity. CSF diversion is frequently required.
In
patients without visual compromise with large tumors filling the third
ventricle, a transcallosal approach to the tumor using a subchoroidal
approach to the third ventricle has been a very useful technique for
removing large volumes of tumor. In some cases the biological effect of
resecting these tumors can lead to its stabilization or even involution
and occasionally to complete disappearance. Patients with an optic glioma
and neurofibromatosis type 1 have a better prognosis than those who do
not have neurofibromatosis.
Adjuvant
therapy:
The
role of adjuvant therapy in the treatment of optic pathway tumors remains
controversial. The young age of the patients considerably limits the use
of radiotherapy but none the less, for posterior tumors, irradiation may
be effective in adults with a 75% 10 year relapse-free survival.
Side-effects of radiation therapy include endocrine dysfunction,
secondary malignancy and an increased risk of developing moyamoya
phenomenon especially in the setting of NF-1. This is particularly true
in children younger than 5 years of age. Patients with an optic glioma
and neurofibromatosis type 1 have a better prognosis than those who do
not have neurofibromatosis.
Of
the chemotherapeutic agents available, the nitrosourea-based cytotoxic
regiments have been shown to result in symptomatic improvement or
stabilization. Certainly, there is a role for chemotherapy in those
patients with progressive disease who are too young to receive
radiotherapy. Good outcome have been reported with chemotherapy. Patients
with an optic glioma and neurofibromatosis type 1 have a better prognosis
than those who do not have neurofibromatosis. More recently, carboplatin
has been reported to be effective in arresting growth in progressive
optic gliomas but a larger trial is required to substantiate this.
|
